

В структурном типе циркона ZrSiO4 (пр. гр. симметрии I41/amd, Z = 4, c/a ≈ 0,9) кристаллизуется большое число соединений с общей формулой ABO4. Данные соединения наряду с уже известными областями применения как природных, так и синтетических фаз представляют теоретический и практический интерес как матрицы для создания материалов с особыми физическими свойствами путем изо- и гетеровалентных замещений в катионной части кристаллической структуры. На основе циркона могут быть также получены материалы, перспективные для утилизации радиоактивных элементов [1].
В структурах типа циркона катионы сорта B находятся в центре тетраэдров {BO4}, которые не связаны между собой общими атомами кислорода и окружают катионы сорта A с образованием вокруг них координационного полиэдра – додекаэдра Хорда (рис. 1, а) Каждый из тетраэдров {BO4} окружен шестью додекаэдрами {AO8} (рис. 1, б).
В соответствии с требованием электронейтральности формульной единицы суммарный заряд катионной части должен быть равен 8, что реализуется в следующих комбинациях зарядов катионов сортов A и B: + 5 и + 3, + 4 и + 4, + 3 и + 5, + 2 и + 6. Согласно концепции валентностей связей [2] сумма валентностей связей в первой координационной сфере для каждого иона должна быть равна абсолютной величине его формального заряда (степени окисления). Расчет валентностей связей осуществляют по получившей наибольшее распространение экспоненциальной зависимости [2]
s = exp[(R0 – d)/b],
где R0 и b – табулированные эмпирические константы, d – межатомное расстояние катион–анион. Расстояния B–O в тетраэдрах {BO4} равны между собой, поэтому валентность связи B–O приближенно равна теоретическому значению n/4, если заряд катиона сорта B равен n. Так как атомы кислорода в структурах типа циркона контактируют с одним катионом сорта B и с двумя катионами сорта A, то остаток валентности связи 2 – n/4 должен распределяться между двумя связями A–O таким образом, чтобы сумма валентностей связей атома кислорода была равна 2, а сумма валентностей связей атома A равнялась его заряду. Для теоретически рассчитанных структур можно получить практическое совпадение рассчитанных и табличных значений зарядов ионов [3, 4]. При расчетах по экспериментальным данным идеальные совпадения довольно редки. В качестве критерия отклонения табличных значений зарядов ионов от рассчитанных по экспериментальным данным может служить глобальный индекс нестабильности (global instability index) GII [2], рассчитываемый по формуле:
GII = [∑(d2/N)]1/2,
где d – разность между табличным и рассчитанным зарядом для N ионов в независимой части элементарной ячейки. Для стабильных кристаллических структур GII < 0,2.
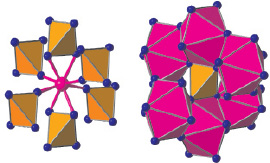
а б
Рис. 1. Фрагменты кристаллической структуры типа циркона: а – окружение катиона A шестью тетраэдрами {BO4}; б – окружение тетраэдра {BO4} шестью полиэдрами {AO8};
В настоящей работе проведено моделирование кристаллических структур семейства циркона с различными комбинациями зарядов катионов сортов A и B для оценки возможности теоретического поиска новых фаз, производных от циркона. В процессе моделирования минимизировался предложенный в [3] функционал
Ф = ∑(ΔZi)2 + ∑[B/(dA–A)12]/2,
где ΔZi – разность между табличным и рассчитанным согласно концепции валентностей связей зарядом иона; dA-A – расстояние анион-анион; B – эмпирическая константа. Расстояниями катион-катион можно пренебречь, так как их учет не вносит существенного вклада в окончательные результаты. Расчет валентностей связей осуществлялся по приведенной ранее экспоненциальной зависимости, параметры R0 и b были взяты из [2]. Использовалось универсальное значение параметра b = 0,37 для всех пар катион-анион.
Проведенные расчеты указывают на удовлетворительное совпадение экспериментальных и теоретических параметров элементарных ячеек модельных структур. Относительные отклонения в параметрах элементарных ячеек, в основном, были менее 1 % и только для структуры хромата кальция CaCrO4 отклонения составили 1,5 % для параметра a и 1,0 % для параметра c (таблица).
Для всех модельных структур были получены низкие значения индекса GII, не превышающие 0,011. Индексы GII рассчитанные по экспериментальным данным находились в пределах от 0,03 до 0,135, что свидетельствует о корректности применения концепции валентностей связей для кристаллохимического анализа рассматриваемых структур.
Результаты моделирования кристаллических структур ABO4 типа циркона*
|
Формула |
Источник экспериментальных данных |
Параметры элементарной ячейки |
|||||
|
a, Å |
c, Å |
||||||
|
Эксп. |
Теор. |
∆, % |
Эксп. |
Теор. |
∆, % |
||
|
ZrSiO4 |
9005518 |
6,6039 |
6,5450 |
0,9 |
5,9783 |
6,0266 |
0,8 |
|
ThGeO4 |
[5] |
7,2399 |
7,2749 |
0,5 |
6,5425 |
6,5305 |
0,2 |
|
LuPO4 |
9013510 |
6,7989 |
6,8037 |
0,07 |
5,9664 |
6,0006 |
0,6 |
|
NbBO4 |
9012847 |
6,2141 |
6,2474 |
0,5 |
5,4760 |
5,5039 |
0,5 |
|
CaCrO4 |
[6] |
7,222 |
7,3337 |
1,5 |
6,285 |
6,3499 |
1,0 |
Примечание. *В столбце «Источник» указаны литературная ссылка или номер записи в базе данных «Crystallography Open Database».
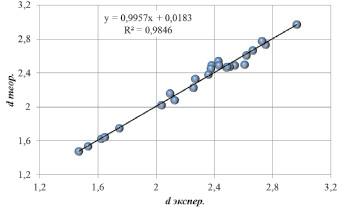
Рис. 2. Основные межатомные расстояния (Å) в структурах типа циркона
Межатомные расстояния A–O, B–O и O–O, рассчитанные по результатам проведенного моделирования, также незначительно отклоняются от экспериментальных значений (рис. 2). Наименьшие расхождения в теоретических и экспериментальных межатомных расстояниях получены для связей B–O в тетраэдрах {BO4} в интервале 1,4–1,8 Å. Это связано с равенством всех четырех расстояний по условиям симметрии тетраэдров {BO4}. Увеличение или уменьшение этих расстояний по сравнению со среднестатистическими расстояниями для известных структур привело бы к отклонению суммы валентностей связей от табличных значения зарядов соответствующих ионов. Для расстояний A–O в додекаэдрах {AO8} в интервале 2,0–2,6 Å расхождения в теоретических и экспериментальных межатомных расстояниях больше, чем для расстояний B–O в тетраэдрах {BO4}. Согласно симметрии додекаэдров {AO8} расстояния A–O разбиваются на две группы неравных расстояний. В этом случае табличное значение заряда катионов сорта A может быть получено при различных комбинациях двух типов расстояний A–O. Проведенные расчеты (рис. 2) указывают на удовлетворительное совпадение теории и эксперимента и для расстояний A–O.
Полученные на основании моделирования известных кристаллических структур типа циркона результаты указывают на перспективность моделирования структур новых фаз, производных от фаз, рассмотренных в настоящей работе.