

Концепция валентностей связей в настоящее время широко используется в кристаллохимическом анализе структур неорганических соединений с ионным типом химических связей [1]. Согласно данной концепции по аналогии с порядком связи для органических соединений вводится понятие валентности связи для взаимодействий катион – анион в структурах неорганических соединений. Сумма валентностей связей для каждого иона должна равняться абсолютному значению его заряда. Зависимость валентности связи от межатомного расстояния не является линейной. Наилучшее совпадение табличных и рассчитанных зарядов ионов получается при использовании экспоненциальной зависимости валентности связи s от межатомного расстояния d:
s=exp[(R0–d)/b],
где R0 и b – табулированные эмпирические параметры. Параметр R0 характеризует конкретное взаимодействие катион – анион. Для параметра b предложено универсальное значение 0.037 нм-1 для всех пар катион – анион, но также используются и индивидуальные значения для каждой пары.
Наряду с оценкой корректности определения кристаллической структуры и идентификации катионов и анионов в спорных случаях применение концепции валентностей связей расширилось до энергетических расчетов [2] и моделирования кристаллических структур [3, 4]. На современном этапе развития компьютерной техники и программного обеспечения по массивам данных сумм валентностей связей, рассчитанных для элементарной ячейки кристалла возможно построение трехмерного распределения отдельных или всех ионов в кристалле [5, 6]. Построение поверхностей одинакового уровня сумм валентностей связей дает информацию о характере распределения ионов в кристалле: локальном или не локальном. Не локальное распределение позволяет в структурах определить каналы проводимости или пути миграции ионов в процессе ионной проводимости.
В настоящей работе нами получены массивы данных сумм валентностей связей для ряда структур с целью оценки возможности использования поверхностей одинакового уровня не только при определении путей миграции ионов, но также при расшифровке кристаллических структур и анализе фазовых переходов первого рода. В целях удобства сравнения результатов для разных структур рассчитывалась разность Δ суммы валентностей связей и абсолютного значения заряда │z│ соответствующего иона:
Δ = ∑exp[(R0-d)/b] – │z│.
Локализации ионов отвечали в этом случае области около нулевых значений Δ. Для оценки возможности миграции ионов и оценки смещений атомов в результате тепловых колебаний использовались значения Δ с относительными отклонениями 0.1 – 0.2 от нулевого значения. Суммирование при расчете сумм валентностей связей проводилось в пределах 0.5–0.7 нм в целях гарантированного учета ионов противоположного знака в пределах первой координационной сферы. Значения параметров R0 были взяты из [1], использовалось универсальное значение параметра b, равное 0.037 нм-1. Для получения поверхностей одинакового уровня полученные массивы данных сумм валентностей связей обрабатывались с помощью программы VESTA [6].
Проведенные нами расчеты для структуры суперионного проводника AgI (рис. 1а) свидетельствуют о корреляции полученных результатов по распределению ионов Ag+ с высокой ионной проводимостью данного соединения. Поверхности одинакового уровня образуют непрерывное распределение по всему объему кристалла в виде связанных вершинами искаженных тетраэдров. В структуре SrTiO3 (рис. 1б) поверхности одинакового уровня не образуют непрерывного распределения, что свидетельствует о малой вероятности суперионной проводимости данного соединения. Формы поверхностей одинакового уровня указывают на ангармоничный характер тепловых смещений катионов и аниона кислорода. Для катиона Ti4+ – это куб с «вытянутыми» вершинами, для катионов Sr2+ – искаженная за счет удлинений вдоль координатных осей сфера, для анионов O2- – тетрагональная призма с закругленными ребрами и вершинами. Данные результаты коррелируют с известными экспериментальными фактами и свидетельствуют о возможности анализа характера тепловых смещений по известным координатам атомов.
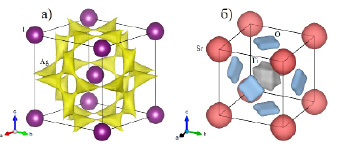
Рис. 1. Поверхности одинакового уровня сумм валентностей связей для катионов Ag+ в структуре AgI (а) и для катионов Sr2+, Ti4+ и анионов O2- в структуре SrTiO3 (б)
Кристаллические структуры PbF2 и PbS имеют гранецентрированные кубические элементарные ячейки с близкой метрикой. В данных структурах катионы свинца образуют плотнейшую кубическую шаровую упаковку. В структуре PbF2 анионы фтора расположены в тетраэдрических пустотах катионной упаковки, а в структуре PbS анионы серы – в октаэдрических пустотах. Нами были проведены расчеты поверхностей одинакового уровня сумм валентностей связей анионов для этих структур с одним и тем же усредненным параметром элементарной ячейки 0.5933 нм. Разница в исходных данных при расчетах заключалась только в параметре взаимодействия катион-анион R0: 0.203 нм в случае структуры PbF2 и 0.2541 нм в случае структуры PbS. Полученные результаты однозначно указывают на заполнение анионами тетраэдрических пустот в первом случае и октаэдрических – во втором (рис. 2). Причем для структуры PbF2 (рис. 2а) проявляется как ангармоничный характер тепловых смещений ионов фтора, так и наличие путей миграции этих ионов, что коррелирует с экспериментальными данными по ионной проводимости PbF2.
В структуре PbS форма поверхностей одинакового уровня близка к сферической (рис. 2б). Локальный характер трехмерного распределения сумм валентностей связей вокруг центров октаэдрических пустот плотнейшей шаровой упаковки катионов Pb2+ указывает на низкую вероятность наличия ионной проводимости в PbS.
Фазовый переход первого рода в структуре ZnS при повышении давления связан с превращением структуры типа сфалерита в структуру типа NaCl. Нами был проведен расчет поверхностей одинакового уровня для фазы ZnS низкого давления и фазы высокого давления с целью локализации ионов серы. При расчетах использовался параметр R0 = 0.209 нм и параметры элементарных ячеек 0.5394 нм для фазы низкого давления и 0.5094 для фазы высокого давления. Полученные результаты (рис. 3) однозначно указывают на структуру сфалерита в случае фазы низкого давления (рис. 3а) и на структуру типа NaCl в случае фазы высокого давления (рис. 3б).
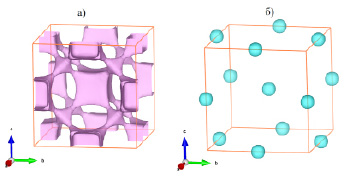
Рис. 2. Поверхности одинакового уровня сумм валентностей связей для анионов F- в структуре PbF2 (а) и для анионов S2- в структуре PbS (б)
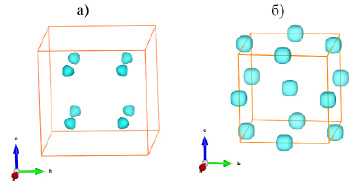
Рис. 3. Поверхности одинакового уровня сумм валентностей связей для анионов S2- в структуре ZnS: а – фаза низкого давления; б – фаза высокого давления
Полученные в настоящей работе результаты указывают на перспективность использования поверхностей одинакового уровня сумм валентностей связей в кристаллохимическом анализе при локализации атомов в структурных исследованиях и при анализе фазовых переходов первого рода.
Библиографическая ссылка
Голубев А.М., Кучина Ю.В., Горячева В.Н., Березина С.Л., Шаповал В.Н., Якушева Е.А. ЛОКАЛИЗАЦИЯ АТОМОВ В КРИСТАЛЛИЧЕСКИХ СТРУКТУРАХ НА ОСНОВЕ РАСЧЕТА СУММ ВАЛЕНТНОСТЕЙ СВЯЗЕЙ // Международный журнал экспериментального образования. 2015. № 8-1. С. 108-110;URL: https://expeducation.ru/ru/article/view?id=7841 (дата обращения: 11.08.2026).